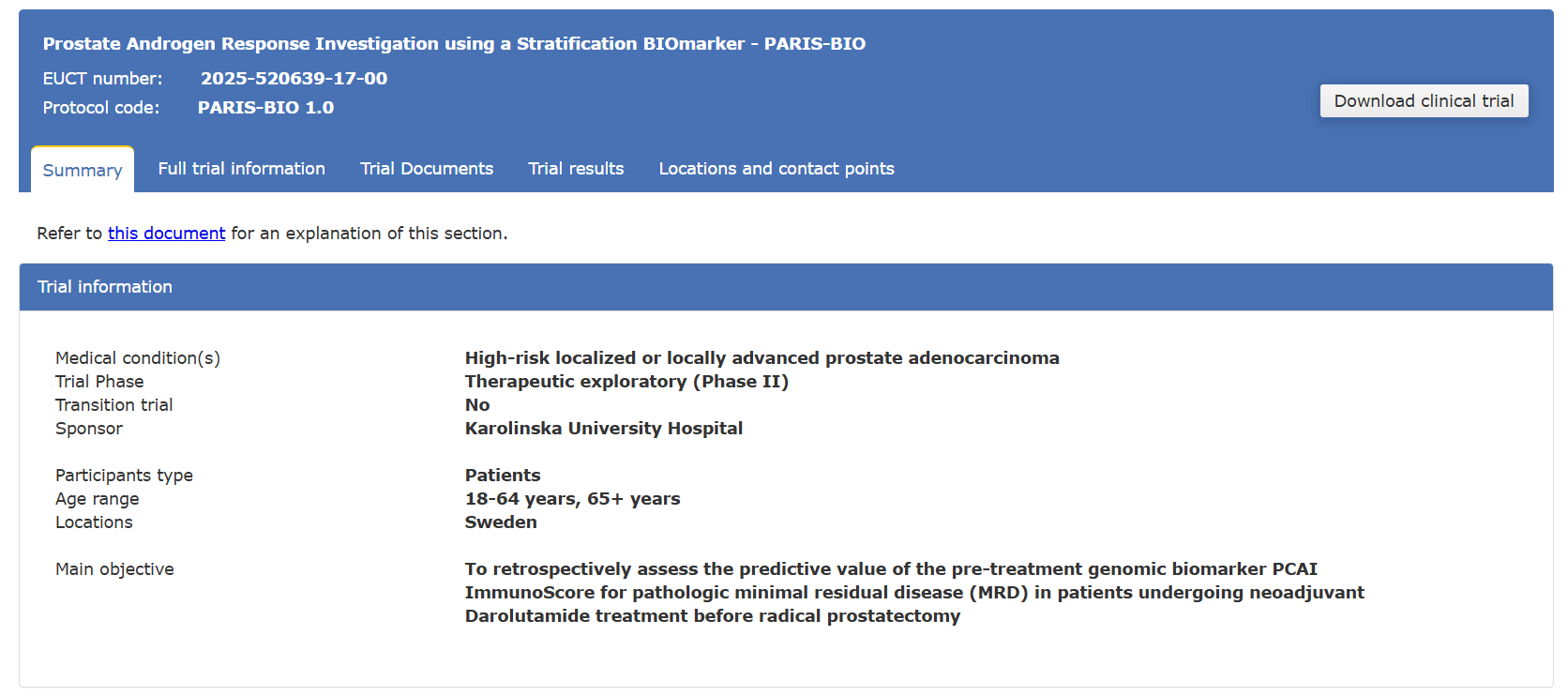
| Metric | What It Indicates | Why It Matters |
| Read Accuracy (Q-score) | Base-calling confidence | Higher accuracy reduces false positives in variant detection |
| Yield (Gb) | Total data generated | Determines if coverage targets are met |
| N50 | Median read length | Reflects ability to resolve complex regions |
| Coverage (X) | Depth across genome | Ensures reliability of detection and assembly |
| Uniformity | Evenness of coverage | Prevents bias and missing regions |
| Contamination Level | Presence of unwanted DNA | Critical for metagenomics and clinical studies |
We use cookies to improve your experience on our site and understand how our content is used. This includes essential cookies for site functionality, as well as optional cookies for analytics and marketing. You can accept all cookies, decline non-essential ones, or manage your preferences below. Declining certain cookies may limit some site features.